https://doi.org/10.29059/cienciauat.v20i1.2041
Desarrollo de modelos QSAR
por redes neuronales para la estimación de potenciales inhibidores de a-glucosidasa
de fenólicos encontrados en matrices alimentarias
Development of QSAR models
using neural networks for the estimation of potential a-glucosidase
inhibitors from phenolics found in food matrices
Modelos QSAR para inhibidores fenólicos
Jesús Jaime
Rochín-Medina*, Hilda Karina Ramírez-Medina, Luis Enrique Barreto-Salazar,
Ángel Ismael Carrillo-Mendivil
*Correspondencia:
jesus.rm@culiacan.tecnm.mx/Fecha de recepción: 6 de julio de 2025/Fecha de
aceptación: 13 de noviembre de 2025/Fecha de publicación: 19 de noviembre de
2023.
Tecnológico Nacional
de México-Instituto Tecnológico de Culiacán, Laboratorio de Microbiología
Molecular y Bioactivos, calle Juan de Dios Bátiz 310 pte., colonia Guadalupe,
Culiacán, Sinaloa, México, C. P. 80220.
Resumen
La
a-glucosidasa es una enzima clave en la digestión de carbohidratos, y su
inhibición representa una estrategia terapéutica para el control de la diabetes
tipo 2. El objetivo del presente estudio fue
desarrollar un modelo QSAR mediante una red neuronal (RNA) y
acoplamiento molecular para estimar la inhibición de a-glucosidasa por
compuestos fenólicos encontrados en matrices alimentarias (CF-MA). La RNA se
construyó para estimar el valor de pIC50 a partir de CF-MA, utilizando distintas configuraciones de neuronas en
la capa oculta. La mejor estimación de pIC50 se logró empleando un
diseño de RNA con 7 neuronas en la capa oculta y dos descriptores moleculares
(SpMin7_Bhm, AATSC5s). El acoplamiento molecular se realizó entre la
a-glucosidasa (isomaltasa) y las móleculas de compuestos fenólicos que
presentaron valores de pIC50 iguales o superiores a la acarbosa (n =
17). El modelo estimó que la naringina (-1.96) y la quercetina
3,4'-O-diglucósido diglucósido (-1.96) presentaron mayor energía libre de afinidad (-10.6 kcal/mol y -10.4 kcal/mol, respectivamente)
frente a la a-glucosidasa, superando al control acarbosa (pIC50 =
-2.00; y energía libre de afinidad -9.8 kcal/mol). La combinación de RNA y acoplamiento molecular permitió
desarrollar una herramienta valiosa para estimar el potencial inhibitorio de
los compuestos fenólicos frente a la enzima de a-glucosidasa.
PALABRAS CLAVE: red neuronal, modelo de predicción, acoplamiento
molecular, compuestos fenólicos, inhibición enzimática.
ABSTRACT
a-Glucosidase is a key enzyme in carbohydrate digestion, and its inhibition
represents a therapeutic strategy for the management of type 2 diabetes. The
aim of this study was to develop a QSAR model using an artificial neural
network (ANN) combined with molecular docking to estimate the inhibitory
activity of a-glucosidase by phenolic compounds found
in food matrices (PC-FM). The ANN was trained to predict pIC50
values using different configurations of hidden-layer neurons. The best
estimation of pIC50 was achieved using an RNA design with 7 neurons in the occult layer and two
molecular descriptors (SpMin7_Bhm, AATSC5s).
Molecular docking was performed between a-glucosidase
(isomaltase) and phenolic compounds that
showed predicted pIC50 values equal to or higher than acarbose (n =
17). According to the model, naringin and quercetin 3,4'-O-diglucoside exhibited the lowest predicted pIC50
values (−1.96) and the highest binding
affinities (−10.6 kcal/mol and −10.4 kcal/mol, respectively),
outperforming the reference compound acarbose (pIC50 = −2.00;
binding affinity = −9.8 kcal/mol). The integration of ANN-based QSAR modeling
and molecular docking provides an efficient tool for screening the inhibitory potential of phenolic compounds against a-glucosidase enzyme.
KEYWORDS: neural network,
predictive model, molecular docking, phenolic compounds, enzyme inhibition.

INTRODUCCIÓN
La diabetes mellitus
es un padecimiento que eleva los niveles de azúcar en sangre (hiper-glucemia) e
índice anormalidades en el sistema metabólico, por lo que representa un desafío
significativo para la salud. Si no se controla, esta condición puede causar
graves daños en diversos órganos y tejidos, incluidos los nervios y vasos
sanguíneos (Yoshikawa y col., 2022). La regulación de la liberación de glucosa
en el tracto intestinal, a partir de los almidones y otros carbohidratos, para
evitar su posterior absorción, puede
lograrse mediante la inhibición de la a-amilasa pancreática, enzima
perteneciente al grupo de las a-glucosidasas que hidroliza el enlace
glucosídico a-1 ® 4. Este mecanismo es
crucial en el control de la hiperglucemia, lo que lo convierte no solo
en un área clave de investigación, sino en una preocupación de salud urgente
que requiere atención e intervención (Poovitha y Parani., 2016)
La Administración de
Alimentos y Medicamentos de los Estados Unidos, aprueba los inhibidores de
a-glucosidasas para el tratamiento de pacientes con diabetes tipo 2. Estos
pueden utilizarse solos, para reducir la hiperglucemia, o en combinación con
otros fármacos antidiabéticos convencionales, en pacientes con niveles
excesivos de glucosa en sangre después de consumir dietas altas en
carbohidratos (Hossain y col., 2020). Por lo tanto, encontrar compuestos
bioactivos naturales inhibidores de a-glucosidasas es una alternativa para
coadyuvar a atender este problema de salud.
La enzima isomaltasa
es otra enzima perteneciente al grupo de las a-glucosidasas, presente en muchos
organismos, incluyendo el ser humano (isomaltasa intestinal), en el que es parte esencial del sistema digestivo. Es responsable de hidrolizar los enlaces glucosídicos
a-1 ® 6 presentes en la amilopectina, oligosacáridos y dextrinas, ayudando a
liberar moléculas de glucosa que son absorbidas en el intestino delgado (Sim y
col., 2010). En consecuencia, la inhibición de la isomaltasa puede reducir la
tasa de digestión de carbohidratos y la absorción de glucosa, disminuyendo los
niveles de azúcar en sangre (Chen y col., 2020). La inhibición de esta enzima,
de origen microbiano, se usa regularmente en estudios de inhibición por su
facilidad de obtención y disponibilidad comercial. Adicionalmente, los
inhibidores de isomaltasas tienen la capacidad de inhibir a otras enzimas del
grupo de las a-glucosidasas, incluyendo las
enzimas pancreáticas humanas, por su similitud estructural (Sim y col.,
2010).
Las matrices
alimentarias y los residuos agro-industriales son
fuentes potencialmente valiosas para incorporarlas en nuevos productos
alimenticios o para extraer de ellas compuestos bioactivos. Diversos estudios
destacan la riqueza y diversidad de los compuestos fenólicos en fuentes vegetales, sus métodos de extracción, así como
su potencial para aplicaciones funcionales y nutracéuticas (Manach y col.,
2004; Wang y col., 2020). Una de las bases de datos con mayor número de
compuestos fenólicos disponibles es Phenol-Explorer, en esta se reúne
información detallada sobre los compuestos
fenólicos contenidos en alimentos, sus formas químicas,
biodisponibilidad y metabolismo (Neveu y col., 2010; Rothwell y col., 2013). Su
uso facilita la identificación de perfiles fenólicos en matrices alimentarias,
así como la selección de compuestos individuales, lo que permite su integración
en análisis in silico, mediante la integración de otras bases de
datos moleculares, como ChEMBL, PubChem
y otras plataformas especializadas (Rothwell y col., 2013; Zdrazil y col.,
2024).
El potencial de los
compuestos fenólicos de matrices alimentarias (CF-MA) en la inhibición
enzimática ha sido un tema de interés, con estudios que atribuyen un efecto
antihiperglucémico a su actividad inhibidora sobre la enzima a-glucosidasa
(Salazar-López y col., 2020; Alongi y col., 2021; Doungwichitrkul y col., 2024). Sin embargo, debido a la no comercialización
de algunos CF-MA, solo unos pocos se han
probado in vitro. Por lo tanto, el uso de alternativas computacionales
para evaluar el potencial de dichos compuestos antes del análisis in
vitro e in vivo es una dirección prometedora en este campo de
investigación (Pinzi y col., 2019).
Una estructura química
se correlaciona cuantitativamente con la actividad biológica o la reactividad
química mediante el uso de descriptores moleculares, que capturan propiedades
fisicoquímicas, topológicas y electrónicas de la molécula. Estas relaciones
pueden modelarse matemáticamente mediante
técnicas como el método de relación estructura-actividad cuantitativa
(QSAR, por sus siglas en inglés: Quantitative Structure-Activity
Relation-ship), permitiendo predecir la actividad biológica a partir de la
estructura (Patra y Chua 2010; Hung y Gini 2021). Estos modelos permiten
identificar patrones en grandes volúmenes de datos químicos previos al
desarrollo de ensayos biológicos exhaustivos, lo cual representa una ventaja
significativa en términos de tiempo, costo y eficiencia (Cherkasov y col.,
2014). Sin embargo, la construcción de modelos QSAR robustos implica un flujo
de trabajo que abarca múltiples etapas, las cuales incluyen la preparación y
curación de datos, detección de valores atípicos, balanceo de conjuntos de
datos, una rigurosa validación interna y externa, así como el establecimiento
de dominios de aplicación para asegurar confiabilidad en las predicciones del
modelo al predecir nuevos compuestos (Tropsha, 2010).
El progreso de la
informática en los últimos años, ha llevado al aumento
del uso de técnicas de inteligencia artificial para resolver problemas
relacionados con la estimación de variables en procesos de diferentes campos de
estudio (Gupta y col., 2021; Javaid y col., 2022). Una de estas técnicas es el
uso de redes neuronales artificiales (RNA), que crean modelos de estimación a
partir de datos obtenidos experimentalmente
(Chapman y col., 2022).
Las RNA se ha
convertido en una herramienta valiosa en algunas investigaciones porque modelan
el comportamiento de un proceso físico, fisiológico, químico y biológico, que
puede ser complejo de representar utilizando modelos matemáticos (Taiwo y col., 2022). Por esta razón, es posible crear un modelo con RNA
utilizando datos experimentales. Además, se han desarrollado modelos QSAR
asistidos por RNA para estimar actividad biológica, como la mutagenicidad
bacteriana, inhibición enzimática relacionadas a enfermedades mentales y
cardiovasculares, entre otras (Patra y Chua 2010; Hung y Gini, 2021; Xu, 2022;
El-fadili y col., 2023).
El objetivo de este
estudio fue desarrollar un modelo QSAR asistido por RNA, complementado con
estudios de acoplamiento molecular, con el fin de estimar la capacidad
inhibidora de compuestos fenólicos presentes en matrices alimentarias, frente a una enzima a-glucosidasa
isomaltasa microbiana.
MATERIALES Y MÉTODOS
Análisis
discriminante sobre descriptores moleculares contra la actividad inhibidora
de la a-glucosidasa
Se optimizó la
respuesta del modelo de RNA mediante un análisis discriminante de los
descriptores moleculares y la actividad inhibitoria de la a-glucosidasa. Para
ello, se consideraron 35 compuestos reportados en la base de datos ChEMBL
(identificador de bioactividad de ensayos: CHEMBL1152716) (Zdrazil y col.,
2024; European Bioinformatics Institute, 2025). Este conjunto de compuestos,
con sus respectivos valores experimentales de IC50 contra
a-glucosidasa microbiana (isomaltasa de Saccharomyces
cerevisiae), permitió determinar los descriptores moleculares utilizados
como datos de entrada en el diseño de la RNA.
Los descriptores
moleculares (1D y 2D) se obtuvieron utilizando el software PaDEL-Descriptor,
versión 2.21, a partir de la Especificación
Simplificada de Entrada Molecular Lineal (SMILES, por sus siglas en
inglés: Simplified Molecular Input Line Entry System), tomando en cuenta
aproximadamente 1 444 descriptores con
distintas características, entre ellos algunos comúnmente usados en QSAR
como LogP y MR (Daoui y col., 2022). Posteriormente, se eliminaron todas las
columnas que contenían valores nulos o constantes (columnas con más del 50 % de
este tipo de datos) para evitar errores
potenciales en las operaciones de matrices. Después de este proceso de
filtrado, se realizó el análisis de correlación
múltiple de Pearson y covarianza con todos los descriptores moleculares
restantes, seleccionando sólo aquellos que no tuvieran codependencia
estadística.
El número óptimo de
descriptores y su contribución a la
variabilidad de los datos (considerando aquellos que explicaran al menos
el 80 % de la variabilidad total) se obtuvo por un análisis de componentes
principales (ACP). Se redujo la multicolinealidad en el modelo por medio de un análisis de conglomerados permitiendo
clasificar los descriptores en familias
según su similitud. Dentro de cada grupo, se seleccionó el descriptor con la
mayor correlación con la actividad inhibitoria (IC50, que
representa la concentración molar -M- que inhibe un efecto al 50 %), eliminando
aquellos que fueran altamente
correlacionados entre sí y procurando la representatividad de las familias
de datos. Todos los análisis
estadísticos fueron realizados utilizando el
software estadístico Minitab® (Minitab Statistical Software), versión 21.
Modelo QSAR con RNA
La RNA utilizó
Regularización Bayesiana para predecir el
valor de pIC50 (-log IC50) (Zerroug y col., 2021)
a partir del conjunto de datos de CF-MA
obtenido de la base de datos Phenol-Explorer (Neveu y col., 2010;
Phenol-Explorer, 2025), con un total
de 501 fenoles disponibles en la plataforma.
La
RNA se construyó a partir de descriptores moleculares mediante el análisis
discriminante, el cual consideró la correlación
de Pearson (r), el gráfico de sedimentación
(que permitió identificar los componentes con mayor varianza explicada) y el
análisis de conglomerados; dichos descriptores mostraron una correlación
significativa (P ≤ 0.05) frente a la actividad inhibidora de la enzima a-glucosidasa
(pIC50) obtenida de la base de datos ChEMBL.
La RNA se construyó
utilizando el 80 % de los datos para el entrenamiento y el 20 % restante para
pruebas y validación. Se probaron cinco modelos con un número diferente de
neuronas en la capa oculta (3, 5, 7, 9 y 15 neuronas). Estos modelos se
desarrollaron en la plataforma MATLAB utilizando la herramienta nftool.
El error cuadrático medio (MSE, por sus
siglas en inglés: Mean Squared Error) se usó para medir la precisión de la RNA,
mientras que el valor de R se empleó para cuantificar la relación entre
los valores experimentales de pIC50 y los valores predichos por la
RNA, permitiendo así evaluar la capacidad predictiva del modelo. El modelo de
RNA seleccionado se utilizó para predecir el valor de pIC50 de todos
los compuestos fenólicos presentes en la base de datos. Posteriormente, se
seleccionaron los compuestos con un pIC50 superior al de la acarbosa
(usada como control positivo) para proceder con el acoplamiento molecular.
Acoplamiento molecular
Se realizaron estudios
de acoplamiento molecular entre los ligandos (CF-MA obtenidos de
Phenol-Explorer) e isomaltasa de Saccha-romyces cerevisiae (PDB 3AJ7),
una a-glucosidasa microbiana con un 72 % de homología con la isomaltasa humana
(Yue y col., 2017) por lo que su uso como modelo estructural resulta adecuado
para la evaluación comparativa de potenciales inhibidores de a-glucosidasas, ya
que la inhibición observada sobre una a-glucosidasa suele ser compatible con la
inhibición potencial de otras enzimas homólogas (Poovitha y Parani, 2016). Para
este análisis, se seleccionaron únicamente aquellos CF-MA con valores de pIC50
mejores al control (acarbosa) y que mostraran interacción con al menos uno de
los residuos catalíticos reportados para a-glucosidasa: Asp215, Glu277 y Asp352
(Yamamoto y col., 2010).
Las
estructuras de los ligandos se obtuvieron de PubChem
(National Library of Medicine, 2025); la estructura proteica de la isomaltasa
(PDB 3AJ7) se obtuvo del Banco de Datos de Proteínas
RCSB (RCSB Protein Data Bank, 2025). La preparación de los archivos para
el acoplamiento se realizó mediante la interfaz de AutodockTools v1.5.6, donde
se añadieron cargas de Gasteiger; también se definieron los residuos
catalíticos y se configuraron las cajas de
búsqueda (Grid box). Las energías de afinidad (Vina score) se procesaron
usando el software libre AutoDock, versión
Vina v1.1.2
Se llevó a cabo el
acoplamiento molecular rígido, manteniendo la estructura de la proteína
completamente fija y utilizando un Grid box
de 27 × 27 × 27 con un espaciado de 0.375 Å, con cada uno de los
ligandos preparados. Posteriormente, se realizó el acoplamiento molecular
flexible, seleccionando únicamente los compuestos con mayores valores de
energía de afinidad que superaron al del control (acarbosa). Para este caso se seleccionaron únicamente a los residuos catalíticos (Asp215, Glu277 y Asp352) como
flexibles, ajustando también el tamaño del Grid box para cubrir de forma
precisa la zona activa. Las interacciones resultantes de cada análisis se
visualizaron mediante Discovery Studio
Visualizer 2021 Client, en formato 2D.
Finalmente,
los compuestos con mejor desempeño en estas etapas fueron sometidos a un acoplamiento
molecular 3D utilizando el software AIDDISON™ (versión 24.0.5.0; plataforma en
línea desarrollada por Sigma-Aldrich)
(Rusinko y col., 2024), seleccionando la caja de búsqueda
correspondiente al sitio activo (Asp215, Glu277 y Asp352), además de incluir
moléculas de agua y los iones metálicos
presentes en la enzima, también se empleó a la acarbosa como control.
RESULTADOS Y DISCUSIÓN
Análisis
discriminante sobre descriptores moleculares contra la actividad inhibidora
de la a-glucosidasa
La eliminación de los
datos inconsistentes o duplicados de los 35 compuestos iniciales obtenidos de
la base de datos de ChEMBL, permitió obtener 30 compuestos finales para el
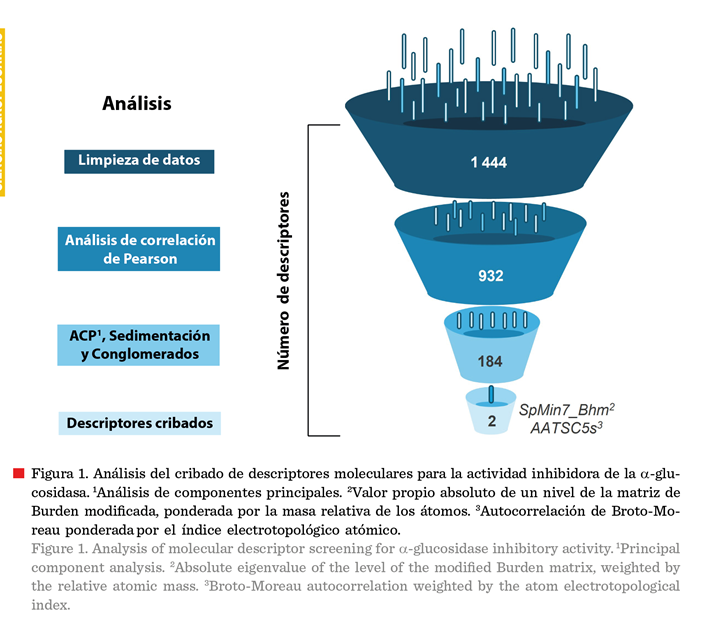
entrenamiento. El flujo de trabajo para cada una de las etapas del análisis está
representado en la Figura 1, mostrando el número de descriptores resultantes y
el análisis aplicado para cada etapa. Los 1 444 descriptores moleculares
inicialmente obtenidos para cada compuesto, se redujeron a 932 descriptores después del proceso de limpieza (eliminación
de duplicados y de datos incompletos o constantes); mediante el análisis de
correlación de Pearson entre los descriptores y el valor de pIC50,
se seleccionaron aquellos descriptores con un coeficiente de correlación (valor
r) menor a -0.7 (para correlaciones negativas) y superior a 0.7 (para
correlaciones positivas), un valor considerado alto para correlaciones y/o colinealidades (Simeon y Jongkon,
2019), resultando un total de 184 descriptores depurados.
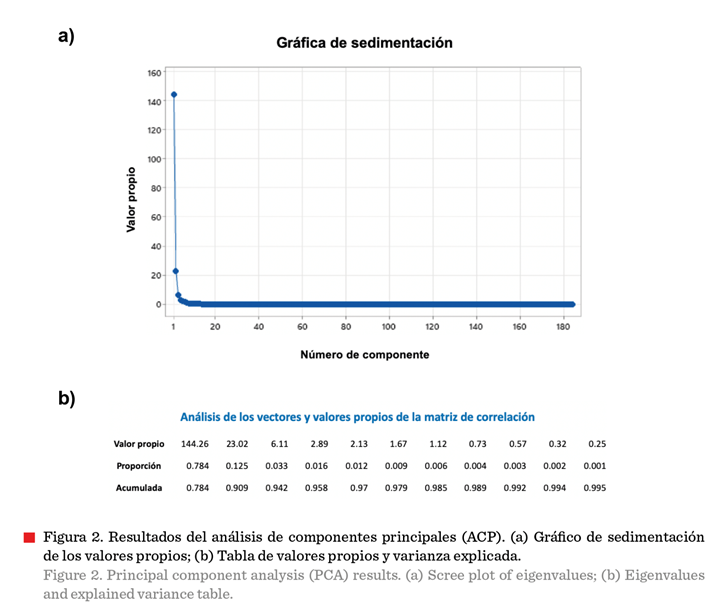
El ACP a través del
análisis de sedimentación (Figura 2) indicó que 2 descriptores permiten
explicar al menos un 90 % de la variabilidad total, lo que sugiere que es
posible utilizarlos para representar el modelo sin una pérdida significativa de
precisión (80 %). Los resultados preliminares mostraron que el descriptor
SpMin7_Bhm, presentó la mayor correlación positiva (r = 0.89) y AATSC5s la
mayor correlación negativa (r = -0.87) contra la variable de respuesta (pIC50)
con valor significativo (P ≤ 0.001). Una correlación positiva indica una relación directa con el pIC50 (incremento),
mientras que una negativa señala una relación inversa (disminución).
Este contraste es útil porque aporta información complementaria y mejora la robustez del modelo (Minitab Support, 2023).
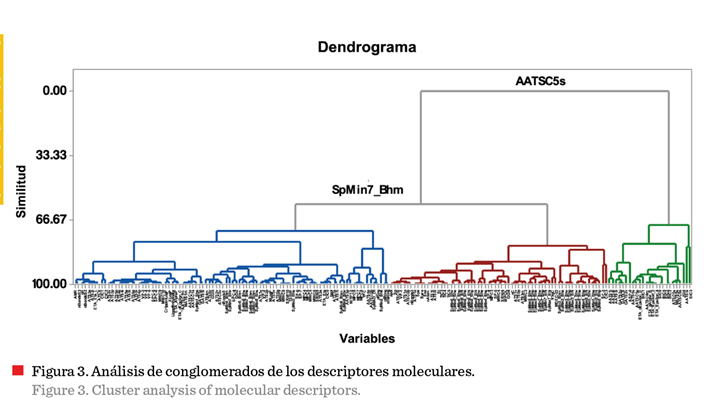
El
análisis de conglomerados (Figura 3) mostró que los descriptores SpMin7_Bhm y
AATSC5s presentaron una distancia euclidiana de 4.78, lo que indica
una baja covariabilidad y los vuelve adecuados para la construcción del modelo,
por lo que fueron seleccionados como las variables de entrada para diseñar el modelo de RNA.
El descriptor
SpMin7_Bhm se basa en propiedades topológicas y representa la energía mínima
estandarizada de Burden en el séptimo orden,
capturando cómo la distribución de la densidad electrónica y la
conectividad del esqueleto molecular pueden influir en la afinidad por el sitio
activo de la enzima (Burden, 1989; Todeschini y Consonni, 2009). Por su parte,
AATSC5s pertenece a la familia de descriptores autocorrelativos y refleja la
suma de cargas parciales ponderadas por la distancia topológica en la quinta
posición, proporcionando información sobre cómo la distribución de carga a lo
largo de la molécula puede facilitar o dificultar interacciones electrostáticas
con residuos catalíticos (Todeschini y Consonni, 2009; Yap, 2011). Diversos
estudios mencionan la importancia de los descriptores del tipo topológico, ya
que se les ha asociado con actividad
biológica, debido a la distribución de la carga y cómo dichas
características son cruciales para ensayos de ese tipo (Gálvez y col., 2002;
Singh y col., 2009; Emonts y col., 2023). Por lo tanto, moléculas con valores
específicos de estos descriptores pueden
mostrar mayor o menor afinidad por la enzima, alterando su capacidad
inhibitoria.
Algunos estudios
destacan la importancia de utilizar descriptores como LogP y MR en ensayos QSAR (Daoui y col., 2022; El-fadili y
col., 2023), sin embargo, en este ensayo no mostraron una correlación
significativa con la actividad inhibitoria de la a-glucosidasa, por lo tanto,
no fueron incluidos en el modelo, lo que resalta la importancia de seleccionar
descriptores específicos y representativos para cada conjunto de datos.
Modelo QSAR con RNA
Los valores estimados
de los cinco modelos construidos inicialmente con 3, 5, 7, 9 y 15 neuronas en
la capa oculta para la estimación del pIC50, se compararon con los
valores de entrenamiento (Tabla 1). Los valores de MSE y R fueron calculados
para cada modelo, y los criterios de selección del modelo se basaron en el MSE
más bajo y un valor R cercano a 1 (Zerroug y col., 2021). Los modelos con 5, 7
y 9 neuronas presentaron un MSE más bajo y un valor R más cercano a 1 durante
la etapa de entrenamiento en comparación con
los modelos con 3 y 15 neuronas. Sin embargo, al comparar los valores de
MSE y R del modelo con 7 neuronas con el modelo con 9 neuronas, se presentó una
diferencia de 0.001 en el MSE y 0.013 en el valor R en la etapa de
entrenamiento, mostrando un rendimiento ligeramente superior el modelo con 9
neuronas. No obstante, en la fase de prueba, el modelo con 7 neuronas reportó
un mejor desempeño, con un MSE más bajo y un valor de R más cercano a 1 (Figura
4).
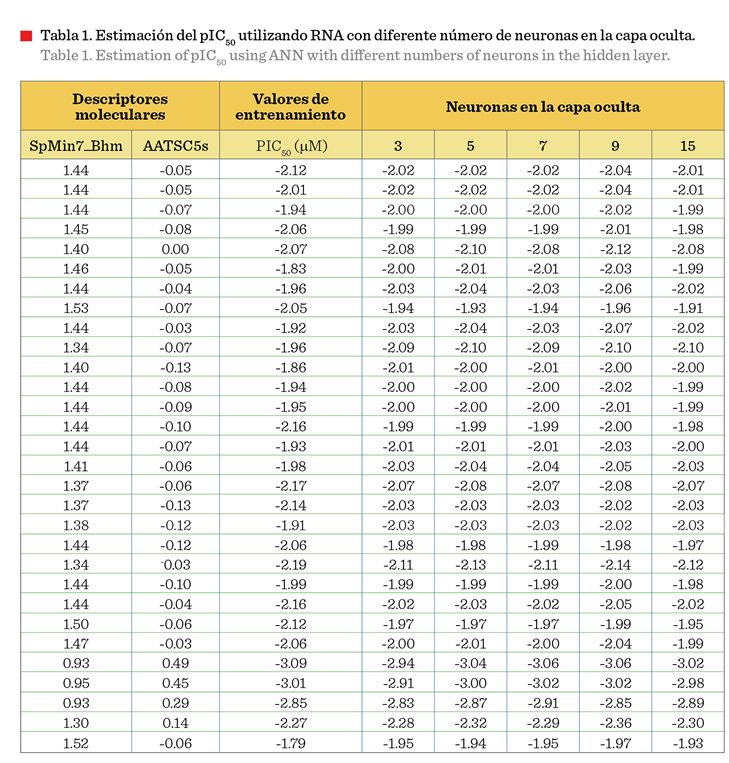
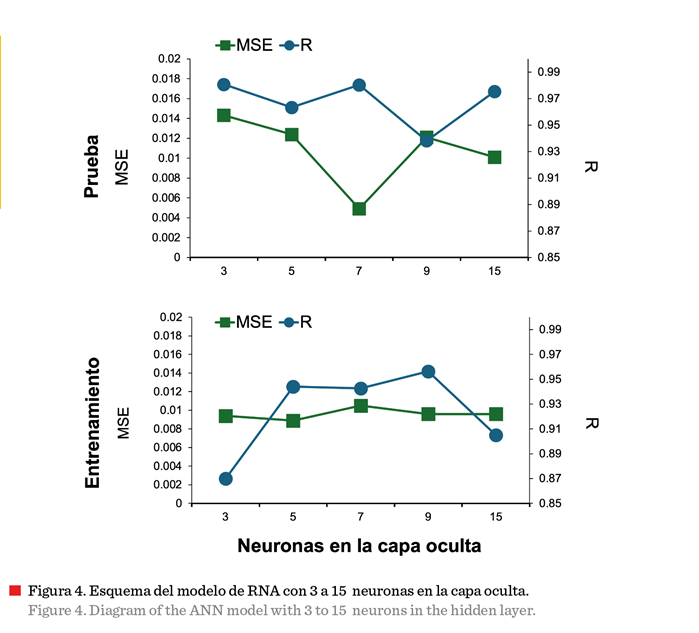
Los resultados de
prueba mostraron una mejora en los valores de
MSE y R del modelo de 7 neuronas en comparación con los modelos con 3,
5, 9 y 15 neuronas, que no reportaron mejora (presentaron valores de MSE más
altos y valores R más alejados de 1). El modelo estimado con 7 neuronas se
ajustó a los valores de entrenamiento (Figura 5a). La evaluación de los
resultados de prueba (Figura 5b) mostró que la línea de regresión se ajusta a
los datos, indicando que el modelo puede predecir datos previamente no observados
y manejar información general desconocida.
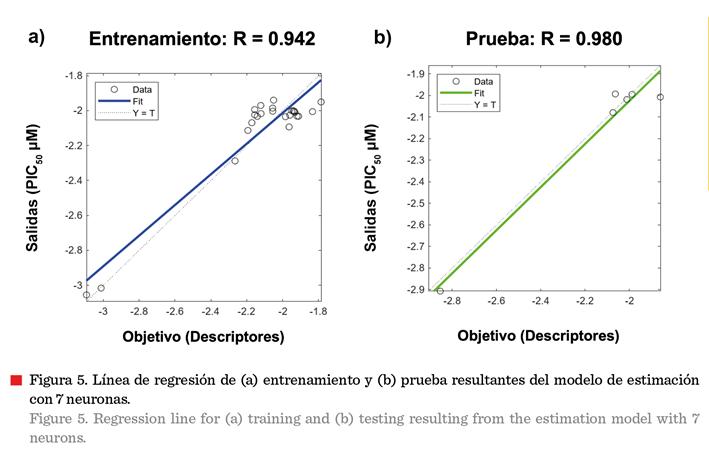
El modelo con 7
neuronas en la capa oculta fue seleccionado para estimar el pIC50,
resultando en un MSE de 0.010 5 y un valor R de 0.942 6 en el entrenamiento
(Figura 5a), valores que indican una alta eficiencia predictiva. Estos valores
fueron mayores en la prueba, obteniéndose un MSE de 0.004 9 y un valor R de
0.980 3 (Figura 5b). La RNA fue construida con los dos descriptores moleculares
seleccionados como variable de entrada (SpMin7_Bhm, AATSC5s), una capa oculta
de 7 neuronas y 1 salida (pIC50). La estructura de la RNA
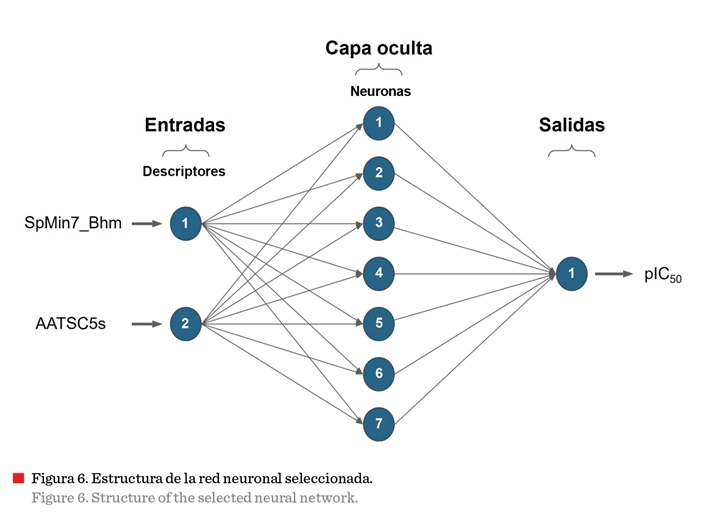
seleccionada se muestra en la Figura 6.
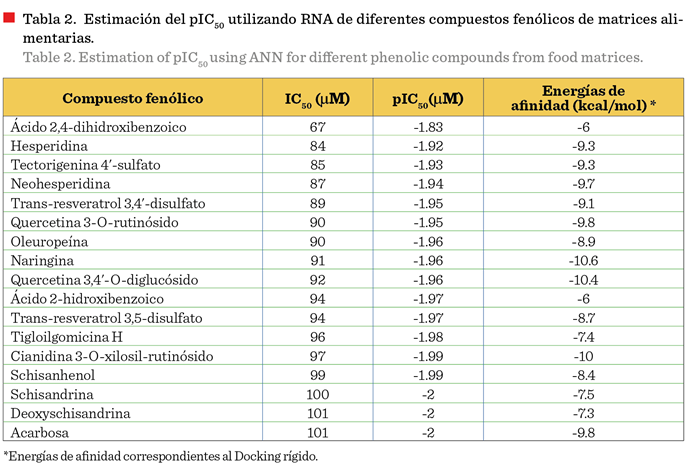
El
cribado se realizó con aquellos compuestos con valores predichos de pIC50
superiores al de la acarbosa, lo que sugiere un posible potencial inhibitorio
(Tabla 2). Entre esos compuestos destacan diversos ácidos fenólicos y
flavonoides, lo que concuerda con estudios previos, que han reportado a dichos
compuestos como posibles inhibidores naturales de a-glucosidasa;
por ejemplo, el ácido benzoico ha mostrado una inhibición significativa in
vitro (45 mM), mientras que flavonoides como el kaemferol (8.9 mM) y la
quercetina (77.4 mM), han demostrado potencial inhibitorio in silico (Guan y col., 2022; Şöhretoğlu y col., 2023).
Los resultados mencionados corresponden a la
etapa de predicción de actividad (pIC50), previa a las evaluaciones cualitativas y al análisis por acoplamiento molecular indicando una tendencia estructural desde esta fase inicial
del análisis. Lo que indica que el uso de herramientas de estimación, como la
RNA, podría ser una herramienta valiosa para predecir la bioactividad
rápidamente de compuestos no evaluados presentes en los alimentos en la
búsqueda de inhibidores naturales de a-glucosidasa. Sin embargo, es necesario
complementar esa estimación con análisis de
acoplamiento molecular, que permita explorar el comportamiento y la afinidad de
los compuestos más prometedores con la enzima.
Acoplamiento molecular
Se llevaron a cabo
distintas técnicas de acoplamiento molecular para determinar la energía de
afinidad entre los compuestos fenólicos (ligandos) cribados de CF-MA y la
isomaltasa, aplicadas de forma secuencial. Se seleccionaron los mejores
complejos enzima-ligando con puntuación (kcal/mol) para los compuestos de mayor
valor estimado de pIC50 en comparación con el control acarbosa
(Tabla 2).
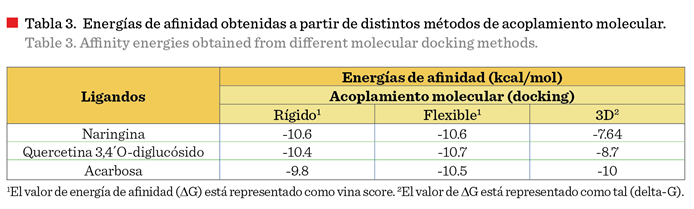
En el acoplamiento
molecular rígido, la naringina exhibió la mayor energía libre de unión (-10.6 kcal/mol), seguida por la quercetina
3,4´O-diglucósido (-10.4 kcal/mol), superando al control acarbosa (-9.8
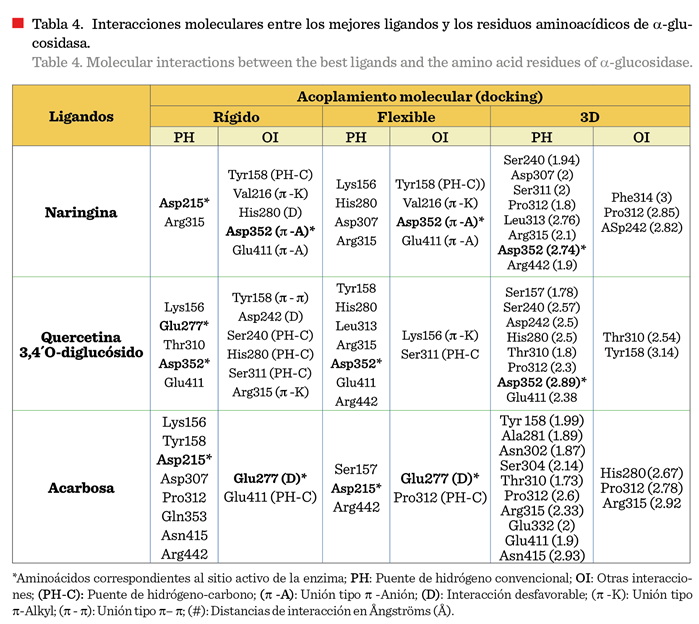
kcal/mol) (Tabla 3). Ambos compuestos interactuaron con los amino-ácidos del sitio activo (Asp215, Glu277, Asp352) de la a-glucosidasa, indicando una inhibición competitiva.
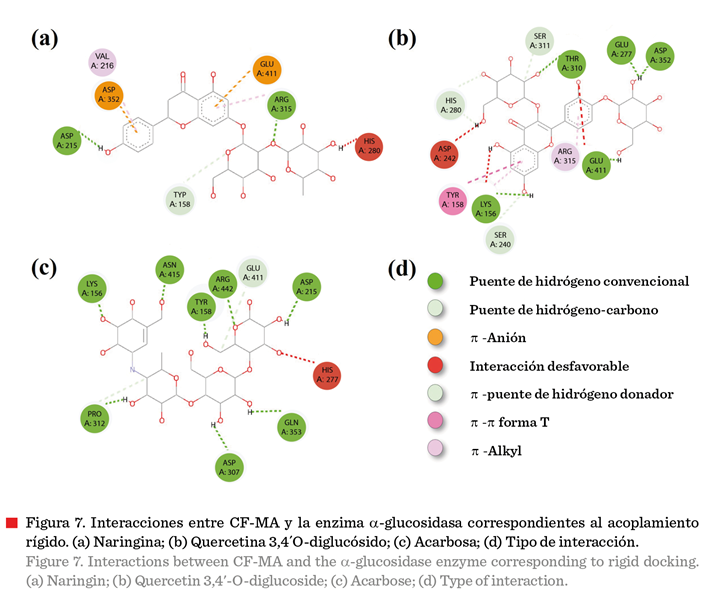
Naringina interactuó con Asp215 mediante un enlace puente de hidrógeno y con
Asp352 mediante una interacción Pi-Anión, mientras que la quercetina
3,4´O-diglucósido interactuó con Glu277 y
Asp352 mediante puentes de hidrógeno (Figura 7). Las interacciones
específicas enzima-ligando de cada uno de los acoplamientos efectuados se
detallan en la Tabla 4.
La parte flexible del
acoplamiento molecular demostró energías libres de unión, similares a la parte
rígida, teniendo como resultado -10.6 kcal/mol con enlaces tipo pi-Anión con el
residuo catalítico Asp352, para naringina, y -10.7 kcal/mol con un enlace tipo
puente de hidrogeno con Asp352 para la quercetina 3,4´O-diglucósido. No
obstante, en ensayo flexible se favorecieron uniones del tipo puente de
hidrógeno con otros residuos en la misma cavidad del sitio catalítico (Tabla 4;
Figura 7).
En el acoplamiento 3D,
realizado en el software AIDDISON, se observaron ligeras diferencias en las
energías de afinidad respecto a los enfoques rígido y flexible, que podría ser
atribuible a variaciones en el cálculo de la energía libre propio del software.
Naringina presentó una afinidad de -7.64 kcal/mol, interactuando con Asp352
mediante puente de hidrógeno, mientras que la quercetina 3,4´O-diglucósido
mostró un valor de -8.7 kcal/mol, interactuando de la misma manera con Asp352.
La acarbosa, en este caso, mantuvo una afinidad ligeramente superior (-10
kcal/mol), sin embargo, no formó ningún enlace en el sitio activo de la enzima,
lo que podría indicar un mecanismo de inhibición menos especifico (inhibición
no competitiva) interactuando solamente en la periferia de la cavidad del sitio activo. Estos resultados muestran que, aunque
la energía libre de afinidad es útil, la especificidad y la naturaleza de las
interacciones con los residuos catalíticos son determinantes para establecer la
actividad inhibitoria real. Por ello, los acoplamientos 3D complementan y
refinan la interpretación obtenida en el acoplamiento rígido y flexible,
resaltando la importancia de considerar tanto la magnitud de la afinidad como
la calidad de las interacciones al evaluar inhibidores potenciales. Aunque los
valores obtenidos para naringina y quercetina 3,4'-O-diglucósido fueron
inferiores a los registrados con los otros enfoques, el acoplamiento 3D permite
incluir moléculas de agua, así como iones metálicos, además de considerar una
mayor flexibilidad de la proteína y del ligando, lo que genera un entorno más
cercano a las condiciones biológicas reales. Por ello, a pesar de que las
energías calculadas sean ligeramente menores, este enfoque podría ofrecer
predicciones más representativas de las interacciones enzimáticas efectivas.
El
acoplamiento rígido permitió realizar un análisis más rápido,
lo que lo vuelve un procedimiento muy eficiente para un primer filtro de compuestos, ya que tanto el ligando como
el sitio activo de la enzima permanecen en conformaciones fijas, reduciendo el
espacio de búsqueda y el costo
computacional. Por su parte, el docking flexible permitió evaluar con
mayor precisión las conformaciones de las estructuras más prometedoras, debido
a que se posibilitan torsiones en la estructura del ligando y de ciertos residuos
del sitio activo, lo que mejora la predicción de la interacción, aunque implica
un mayor tiempo de cálculo y consumo de recursos computacionales. La
combinación de ambos análisis mejora la rapidez inicial, mediante predicción de
la afinidad de unión y la caracterización de las interacciones moleculares,
proporcionando resultados más realistas y biológicamente relevantes (Rosenfeld
y col., 1995; Nirwan y col., 2022).
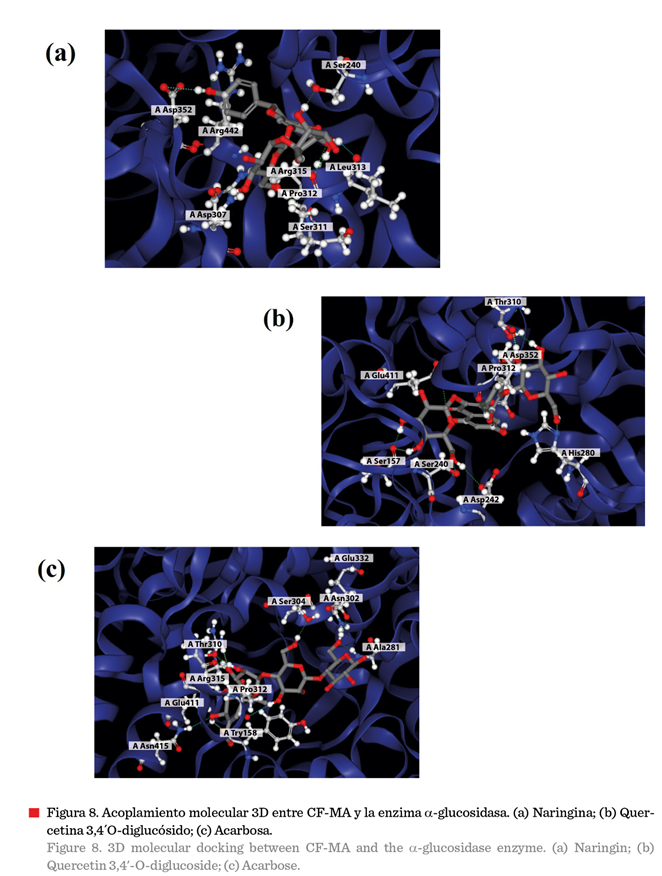
El
software de Sigma-Aldrich, AIDDISON™, versión
24.0.5.0; empleado permitió visualizar los resultados obtenidos, directamente
en la estructura cristalina de la enzima, mostrando cómo el ligando se une al
sitio activo (Figura 8). Mediante esta
visualización 3D no solo es posible confirmar la localización de los
ligandos dentro o fuera del sitio activo y sus distancias, sino también
examinar cómo factores espaciales afectan la estabilidad de la interacción,
aportando así un enfoque complementario y más realista para la evaluación de
potenciales inhibidores (Rusinko y col., 2024). La naringina y la quercetina
3,4'-O-diglucósido fueron los compuestos más destacados, mostrando afinidades elevadas y múltiples interacciones
en todos los análisis de acoplamiento realizados. Este tipo de resultados
permiten establecer posibles comparaciones con resultados experimentales
previamente reportados en la literatura. Por ejemplo, se ha documentado que la
naringina presenta una alta inhibición contra la enzima a-glucosidasa humana in
vitro (IC50= 0.55 mM) y un mayor número de interacciones del
tipo puente de hidrógeno en el ensayo in silico (11 enlaces), en comparación con la acarbosa (IC50 = 108 mM
y 9 enlaces) (Sahnoun y col., 2017). De forma complementaria, otro
estudio reportó un IC50 de 0.174 mmol/L para la naringina (174 mM)
con una energía de unión de −7.6 kJ/mol, superando a los 0.721 mmol/L de
acarbosa (721 mM) (Xiangju y col., 2022).
Los resultados
obtenidos en el presente estudio indicaron que, se estimó un IC50 de
91 mM para la naringina y 101 mM para la acarbosa, utilizando datos de a-glucosidasa microbiana. Esta diferencia
en la inhibición podría deberse al tipo de enzima empleada y al carácter
estimativo del modelo QSAR, cuyo objetivo es facilitar la identificación
temprana de compuestos con potencial terapéutico, acelerando la selección de
candidatos antes de ensayos experimentales. Aun así, los resultados reflejan
correctamente la tendencia observada en la literatura y respaldan el potencial de la naringina como inhibidor de la a-glucosidasa.
En el caso de la
quercetina 3,4'-O-diglucósido, estudios previos reportan una actividad
inhibidora menor frente a la a-glucosidasa, con un valor IC50 de
30.3 mg/mL (48.4 mM), mientras que la acarbosa mostró una mayor inhibición con
un IC50 de 10.1 mg/mL (15.6 mM) (Nile y col., 2021). En contraste
con los resultados obtenidos en el presente estudio, el valor de IC50
predicho de la quercetina 3,4'-O-diglucósido (92 mM) fue superior al de
acarbosa (101 mM), lo que contradice los datos experimentales disponibles. Esta
discrepancia puede atribuirse a las diferencias entre los métodos in silico
e in vitro, ya que los primeros dependen del conjunto de datos y la
parametrización del modelo, mientras que los segundos están condicionados por
la fuente enzimática, el pH y el medio de reacción empleados. Aun así, la
tendencia general de las predicciones concuerda con los comportamientos
relativos observados para otros flavonoides como el kaemferol (Şöhretoğlu y
col., 2023), lo que respalda la capacidad del modelo como herramienta estimativa útil en el cribado y selección
de inhibidores potenciales.
Los
resultados obtenidos mediante la RNA permitieron predecir
los compuestos con mayor probabilidad de inhibir la enzima, mientras que el
acoplamiento molecular validó estructuralmente dichas predicciones al mostrar
interacciones estables en el sitio activo. De este modo, ambos enfoques se
complementan, integrando la predicción
cuantitativa de la RNA con la confirmación estructural del docking.
Algunos autores reportan una relación positiva entre los ensayos de
acoplamiento molecular y los ensayos in vitro (Tolmie y col., 2021; Mehmood y col., 2022) y han enfatizado la
importancia de los ensayos in silico para estimar la inhibición
enzimática, especialmente para la evaluación de nuevas moléculas antes de los
ensayos in vitro e in vivo. Esto respalda el enfoque adoptado en
el presente trabajo, donde las predicciones in silico no solo ofrecieron
resultados coherentes con la literatura, sino que también permitieron
identificar compuestos prometedores para futuros estudios experimentales. Cabe
destacar que la combinación de datos de Phenol-Explorer y ChEMBL constituye un
aspecto innovador del estudio, al integrar información estructural y bioactiva
de compuestos fenólicos, fortaleciendo la capacidad predictiva del modelo y su
aplicabilidad en la identificación de nuevos inhibidores de a-glucosidasa.
Aunque los compuestos identificados ya se habían reportado como inhibidores de
a-glucosidasa, la aportación principal es la validación de un enfoque eficiente
y adaptable a otros sistemas enzimáticos, capaz de priorizar compuestos con alto potencial bioactivo y reducir el
tiempo y recursos necesarios para la evaluación experimental.
CONCLUSIONES
La RNA desarrollada,
configurada con siete neuronas en la capa oculta e implementando los
descriptores SpMin7_Bhm y AATSC5s como variables de entrada, demostró una
elevada capacidad predictiva para estimar la actividad inhibidora frente a la
enzima a-glucosidasa (isomaltosa) (R = 0.9803). La selección de estos
descriptores constituye una contribución técnica relevante, al evidenciar su
eficacia para modelar la inhibición en comparación con descriptores comúnmente
empleados como LogP o MR. Además, el uso de análisis de correlación de Pearson,
componentes principales y conglomerados
permitió reducir los 1 444 descriptores iniciales a dos, de manera
rigurosa y bien justificada, minimizando la multicolinealidad y maximizando la
representatividad de los datos. La combinación del modelado QSAR mediante RNA y
los análisis de acoplamiento molecular
representa un aporte significativo a la
quimioinformática aplicada, al proporcionar un enfoque dual para
predecir y validar la actividad biológica de compuestos fenólicos, reduciendo
la necesidad de ensayos experimentales preliminares y orientando de manera más
eficiente la búsqueda de nuevos inhibidores naturales de a-glucosidasa. Entre
los compuestos evaluados, la naringina y la quercetina 3,4'-O-diglucósido
destacaron como los candidatos más prometedores, al presentar afinidades
superiores a las de la acarbosa tanto en las predicciones del modelo como en
los análisis de docking (rígido, flexible y 3D), lo que refuerza su potencial como inhibidores de interés farmacológico.
El presente diseño metodológico integra
herramientas estadísticas y computacionales de forma estructurada, con
lo que demuestra la capacidad del enfoque
QSAR–RNA–Docking para filtrar compuestos bioactivos de manera eficiente.
Es necesario corroborar experimentalmente los resultados obtenidos por este método y ampliar la base de datos con nuevos compuestos y descriptores, a fin defortalecer la robustez y generalización del
modelo.
Declaración de conflicto de
intereses
Los autores declararon
no tener conflictos de intereses de ningún tipo.
Referencias
Alongi, M., Frías-Celayeta, J. M., Vriz, R., Kinsella, G.
K., Rulikowska, A., & Anese, M. (2021). In vitro digestion nullified
the differences triggered by roasting in phenolic composition and a-glucosidase
inhibitory capacity of coffee. Food Chemistry, 342, 128289. https://doi.org/10.1016/j.foodchem.2020.128289
Burden, F. R. (1989). Molecular identification
number for substructure searches. Journal
of Chemical Information and Computer Sciences,
29(3), 225-227. https://doi.org/10.1021/ci00063a011
Chapman, J., Truong, V. K., & Cozzolino, D. (2022). Artificial
intelligence applied to healthcare and biotechnology. In D. Barh (Ed.), Biotechnology
in Healthcare: Technologies and Innovations (pp. 249- 257). Elsevier. https://doi.org/10.1016/B978-0-323-89837-9.00001-2
Chen, J. G., Wu, S. F., Zhang, Q. F., Yin, Z. P., &
Zhang, L. (2020). a-Glucosidase inhibitory effect of anthocyanins from
Cinnamomum camphora fruit: Inhibition
kinetics and mechanistic insights through in vitro and in
silico studies. International Journal of Biological Macromolecules,
143, 696-703. https://doi.org/10.1016/j.ijbiomac.2019.09.091
Cherkasov, A., Muratov, E. N., Fourches, D., Varnek, A.,
Baskin, I. I., Cronin, M., Dearden, J., Gramatica, P., Martin, Y. C.,
Todeschini, R., Consonni, V., Kuz’Min, V. E., Cramer, R., Benigni, R., Yang,
C., Rathman, J., Terfloth, L., Gasteiger, J., Richard, A., & Tropsha, A.
(2014). QSAR modeling: Where have you been? Where are you going to? Journal
of Medicinal Chemistry, 57(12), 4977-5010. https://doi.org/10.1021/jm4004285
Daoui, O., Mkhayar, K., Elkhattabi, S., Chtita, S., Zgou,
H., & Elkhalabi, R. (2022). Design of novel carbocycle-fused quinoline
derivatives as potential inhibitors of lymphoblastic leukemia cell line MOLT-3
using 2D-QSAR and ADME-Tox studies. RHAZES: Green and Applied Chemistry,
14(0), 36-61. https://doi.org/10.48419/IMIST.PRSM/RHAZES-V14.31152
Doungwichitrkul, T., Damsud, T., & Phuwapraisirisan,
P. (2024). a-Glucosidase Inhibitors from Cold-Pressed Black Sesame (Sesamum
indicum) Meal: Characterization of New Furofuran Lignans, Kinetic Study, and In
Vitro Gastrointestinal Digestion. Journal of Agricultural and Food Chemistry,
72(2), 1044-1054. https://doi.org/10.1021/acs.jafc.3c04159
El-fadili,
M., Er-rajy, M., Imtara, H., Noman, O. M., Mothana, R. A., Abdullah, S.,
Zerougui, S., & Elhallaoui, M. (2023). QSAR,
ADME-Tox, molecular docking and molecular dynamics simulations of novel
selective glycine transporter type 1 inhibitors with memory enhancing
properties. Heliyon, 9(2), e13706. https://doi.org/10.1016/j.heliyon.2023.e13706
Emonts,
J. & Buyel, J. F. (2023). An overview of descriptors to capture protein properties – Tools and perspectives
in the context of QSAR modeling. Computational and Structural Biotechnology
Journal, 21, 3234–3247. https://doi.org/10.1016/J.CSBJ.2023.05.022
European
Bioinformatics Institute (2025). Base de datos ChEMBL. [En línea]. Disponible
en: https://www.ebi.ac.uk/chembl/. Fecha de
consulta: 15 de marzo de 2025.
Gálvez,
J., Garcia, R., Salabert, M. T., & Soler, R. (2002). Charge Indexes. New
Topological Descriptors. Journal of Chemical Information and
Computer Sciences, 34(3), 520–525. https://doi.org/10.1021/CI00019A008
Guan, L., Long, H., Ren, F., Li, Y.,
& Zhang, H. (2022). A Structure—Activity Relationship Study
of the Inhibition of a-Amylase by Benzoic Acid and Its Derivatives. Nutrients, 14(9),
1931. https://doi.org/10.3390/NU14091931/S1
Gupta,
R., Srivastava, D., Sahu, M., Tiwari, S., Ambasta, R. K., & Kumar, P.
(2021). Artificial intelligence to deep learning: machine
intelligence approach for drug discovery. Molecular Diversity, 25(3),
13151360. https://doi.org/10.1007/s11030-021-10217-3
Hossain,
U., Das, A. K., Ghosh, S., & Sil, P. C. (2020). An
overview on the role of bioactive a-glucosidase inhibitors in ameliorating diabetic complications. Food and
Chemical Toxicology, 145, 111738. https://doi.org/10.1016/j.fct.2020.111738
Hung, C. & Gini, G. (2021). QSAR modeling without
descriptors using graph convolutional neural networks: the case of mutagenicity
prediction. Molecular Diversity, 25(3), 1283-1299. https://doi.org/10.1007/s11030-021-10250-2
Javaid, M., Haleem, A., Singh, R. P., & Suman, R.
(2022). Artificial Intelligence Applications for Industry 4.0: A
Literature-Based Study. Journal of Industrial Integration and Management,
7(1), 83-111. https://doi.org/10.1142/S2424862221300040
Manach, C., Scalbert, A., Morand, C., Rémésy, C., &
Jiménez, L. (2004). Polyphenols: food sources and bioavailability. The American Journal of Clinical Nutrition,
79(5), 727-747. https://doi.org/10.1093/AJCN/79.5.727
Mehmood, R., Mughal, E. U., Elkaeed, E.
B., Obaid, R. J., Nazir, Y., Al-Ghulikah, H. A., Al-Ghulikah, H., Naeem, N., Munirah
M. Al-Rooqi, Ahmed, S. A., Wadood- Shah, A. S., Sadiq, A. (2022).
Synthesis of Novel 2,3-Dihydro-1,5-Benzothiazepines as a-Glucosidase
Inhibitors: In Vitro, In Vivo, Kinetic, SAR, Molecular Docking, and QSAR
Studies. ACS Omega, 7(34), 30215-30232. https://doi.org/10.1021/acsomega.2c03328
Minitab
Support. (2023). Interpretar todos los estadísticos y gráficas para
Correlación. Minitab. [En línea]. Disponible en:
https://support.minitab.com/es-mx/minitab/help-and-how-to/statistics/basic-statistics/how-to/correlation/interpret-the-results/all-statistics-and-graphs/
Fecha de consulta: 16 de octubre de 2025.
National
Library of Medicine (2025). PubChem. [En línea]. Disponible en:
https://pubchem.ncbi.nlm.nih.gov. Fecha de consulta: 15 de marzo de 2025.
Neveu,
V., Perez-Jiménez, J., Vos, F., Crespy, V., du-Chaffaut, L., Mennen, L., Knox,
C., Eisner, R., Cruz, J., Wishart, D., & Scalbert, A. (2010).
Phenol-Explorer: an online comprehensive database on polyphenol contents in
foods. Database : The Journal of Biological
Databases and Curation, 2010. https://doi.org/10.1093/DATABASE/BAP024
Nile, A., Gansukh, E., Park, G. S., Kim, D. H., &
Nile, S. H. (2021). Novel insights on the multi-functional properties of
flavonol glucosides from red onion (Allium cepa L) solid waste – In vitro and
in silico approach. Food Chemistry, 335, 127650. https://doi.org/10.1016/J.FOODCHEM.2020.127650
Nirwan, S., Chahal, V., & Kakkar, R. (2022). A
comparative study of different docking methodologies to assess the
proteinligand interaction for the E. coli MurB enzyme. Journal of
Biomolecular Structure & Dynamics, 40(21), 11229-11238. https://doi.org/10.1080/07391102.2021.1957019
Patra, J. C. & Chua, K. H. K. (2010). Neural network
based drug design for diabetes mellitus using QSAR
with 2D and 3D descriptors. The 2010 International Joint Conference on
Neural Networks (IJCNN), 1-8. https://doi.org/10.1109/IJCNN.2010.5596935
Phenol-Explorer
(2025). Base de datos sobre compuestos fenólicos en alimentos. [En línea].
Disponible en: http://phenol-explorer.eu/. Fecha de consulta: 16 de marzo de
2025.
Pinzi, L. & Rastelli, G. (2019). Molecular Docking:
Shifting Paradigms in Drug Discovery. International Journal of Molecular
Sciences, 20(18), 4331. https://doi.org/10.3390/ijms20184331
Poovitha, S., & Parani, M. (2016). In vitro and in
vivo a-amylase and a-glucosidase inhibiting activities of the protein extracts
from two varieties of bitter gourd (Momordica charantia L.). BMC Complementary and Alternative Medicine, 16(S1), 185. https://doi.org/10.1186/s12906-016-1085-1
RCSB
Protein Data Bank (2025). Banco de Datos de Proteínas RCSB. [En línea].
Disponible en: https://www.rcsb.org. Fecha de consulta: 15 de marzo de 2025.
Rosenfeld,
R., Vajda, S., & DeLisi, C. (1995). Flexible docking and
design. Annual Review of Biophysics and Biomolecular Structure, 24(1),
677-700.https://doi.org/10.1146/ANNUREV.BB.24.060195.003333/CITE/REFWORKS
Rothwell, J. A., Perez-Jimenez, J., Neveu, V.,
Medina-Remón, A., M’Hiri, N., García-Lobato, P., Manach, C., Knox, C., Eisner,
R., Wishart, D. S., & Scalbert, A. (2013). Phenol-Explorer 3.0: a major
update of the Phenol-Explorer database to incorporate data on the effects of
food processing on polyphenol content. Database, 2013. https://doi.org/10.1093/DATABASE/BAT070
Rusinko, A., Rezaei, M., Friedrich, L., Buchstaller, H.
P., Kuhn, D., & Ghogare, A. (2024). AIDDISON: Empowering Drug Discovery
with AI/ML and CADD Tools in a Secure, Web-Based SaaS Platform. Journal of
Chemical Information and Modeling, 64(1), 3-8. https://doi.org/10.1021/acs.jcim.3c01016
Sahnoun,
M., Trabelsi, S., & Bejar, S. (2017). Citrus flavonoids collectively
dominate the a-amylase and a-glucosidase inhibitions. Biologia (Poland),
72(7), 764-773. https://doi.org/10.1515/biolog-2017-0091
Sim,
L., Willemsma, C., Mohan, S., Naim, H. Y., Pinto, B. M., & Rose, D. R.
(2010). Structural Basis for Substrate Selectivity in Human
Maltase-Glucoamylase and Sucrase-Isomaltase N-terminal Domains. The Journal
of Biological Chemistry, 285(23), 17763. https://doi.org/10.1074/JBC.M109.078980
Salazar-López,
N. J., López-Rodríguez, C. V., Hernández-Montoya, D. A., & Campos-Vega, R.
(2020). Health Benefits of Spent Coffee Grounds. In Food
Wastes and By-products, 327-351). https://doi.org/10.1002/9781119534167.ch11
Simeon, S. & Jongkon, N. (2019). Construction of Quantitative Structure Activity Relationship
(QSAR) Models to Predict Potency of Structurally Diversed Janus Kinase 2
Inhibitors. Molecules 24(23), 4393 https://doi.org/10.3390/MOLECULES24234393
Singh,
P., Kumar, R., Sharma, B. K., & Prabhakar, Y. S. (2009). Topological
descriptors in modeling malonyl coenzyme A decarboxylase inhibitory activity:
N-Alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypro-pylphenyl) amide derivatives. Journal of Enzyme Inhibition and Medicinal Chemistry, 24(1), 77–85. https://doi.org/10.1080/14756360801915336
Şöhretoğlu,
D., Renda, G., Arroo, R., Xiao, J., & Sari, S. (2023). Advances
in the natural a-glucosidase inhibitors. EFood, 4(5). https://doi.org/10.1002/efd2.112
Taiwo, A. E., Okoji, A. I., Eloka-Eboka, A. C., &
Musonge, P. (2022). The role of artificial neural networks in bioproduct
development: a case of modeling and optimization studies. Current Trends and
Advances in Computer-Aided Intelligent Environmental Data Engineering,
417-431. https://doi.org/10.1016/B978-0-323-85597-6.00007-0
Todeschini,
R. & Consonni, V. (2009). Molecular Descriptors for Chemoinformatics:
Volume I: Alphabetical Listing (Second edition). Wiley-VCH.
Tolmie,
M., Bester, M. J., & Apostolides, Z. (2021). Inhibition of a-glucosidase
and a-amylase by her-bal compounds for the treatment of type 2 diabetes: A
validation of in silico reverse docking with in vitro enzyme assays. Journal of Diabetes, 13(10), 779-791. https://doi.org/10.1111/1753-0407.13163
Tropsha, A. (2010). Best Practices for QSAR Mo-del Development, Validation, and Exploitation. Molecular
Informatics, 29(6-7), 476-488. https://doi.org/10.1002/MINF.201000061
Wang, Z., Li, S., Ge, S., & Lin,
S. (2020). Review of Distribution, Extraction Methods, and Health Benefits of
Bound Phenolics in Food Plants. Journal of Agricultural and Food Chemistry, 68(11), 3330-3343. https://doi.org/10.1021/acs.jafc.9b06574
Xiangju, Z., Yuqin, C., Zhongping,
Y., Qi, L., Jianwei, Z., Daobang, T., Jiguang, C., Xiangju, Z., Yuqin, C.,
Zhongping, Y., Qi, L., Jianwei, Z., Daobang, T., & Jiguang, C. (2022).
Inhibitory Effect of Naringin on a-Glucosidase and Its Mechanism. Science
and Technology of Food Industry, 43(8), 157-164. https://doi.org/10.13386/J.ISSN1002-0306.2021080184
Xu, Y. (2022). Deep neural networks
for QSAR. In A. Heifetz (Ed.), Artificial intelligence in drug design (pp.
233-260). Humana. https://doi.org/10.1007/978-1-0716-1787-8_10
Yamamoto, K., Miyake, H., Kusunoki, M., & Osaki, S.
(2010). Crystal structures of isomaltase from
Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose.
FEBS Journal, 277(20), 4205-4214. https://doi.org/10.1111/j.1742-4658.2010.07810.x
Yap, C. W. (2011). PaDEL-descriptor: An open source
software to calculate molecular descriptors and fingerprints. Journal of
Computational Chemistry, 32(7), 1466-1474. https://doi.org/10.1002/JCC.21
707
Yoshikawa,
T., Mifune, Y., Inui, A., Nishimoto, H., Yamaura, K., Mukohara, S., Shinohara,
I., & Kuroda, R. (2022). Quercetin treatment protects the
Achilles tendons of rats from oxidative stress induced by hyperglycemia. BMC
Musculoskeletal Disorders, 23(1), 563. https://doi.org/10.1186/s12891-02205513-4
Yue, L. M., Lee, J., Zheng, L., Park,
Y. D., Ye, Z. M., & Yang, J. M. (2017). Computational prediction
integrating the inhibition kinetics of gallotannin on a-glucosidase. International
Journal of Biological Macromolecules, 103, 829-838. https://doi.org/10.1016/j.ijbiomac.2017.05.106
Zdrazil, B., Felix, E., Hunter, F., Manners, E. J.,
Blackshaw, J., Corbett, S., de-Veij, M., Ioannidis, H., Lopez, D. M., Mosquera,
J. F., Magarinos, M. P., Bosc, N., Arcila, R., Kizilören, T., Gaulton, A.,
Bento, A. P., Adasme, M. F., Monecke, P., Landrum, G. A., & Leach, A. R.
(2024). The ChEMBL Database in 2023: a drug discovery platform spanning
multiple bioactivity data types and time periods. Nucleic Acids Research, 52(D1),
D1180-D1192. https://doi.org/10.1093/NAR/GKAD1004
Zerroug, E., Belaidi, S., & Chtita, S. (2021).
Artificial neural network-based quantitative structure–activity relationships
model and molecular docking for virtual screening of novel potent
acetylcholines-terase inhibitors. Journal of the Chinese Chemical Society,
68(8), 1379-1399. https://doi.org/10.1002/jccs.202000457